An Introduction
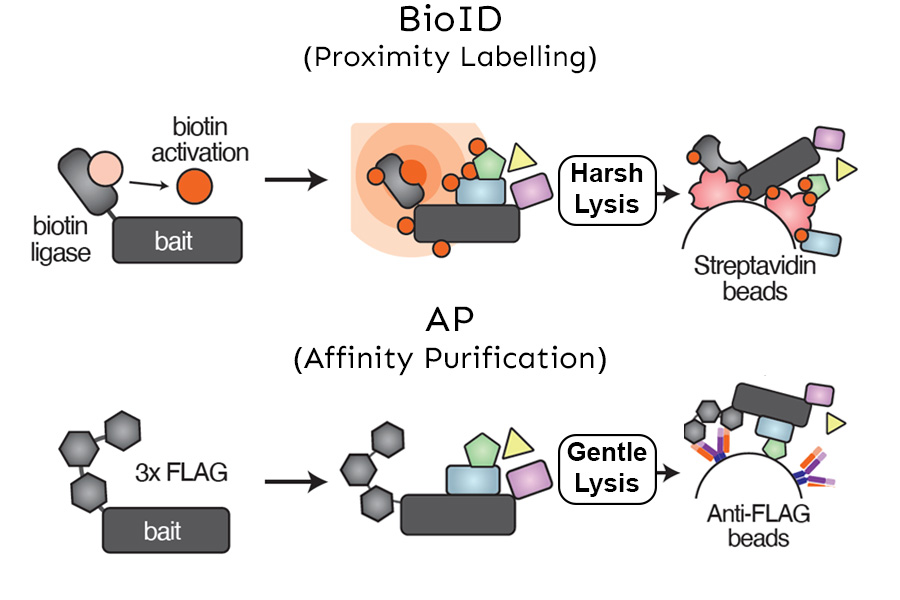
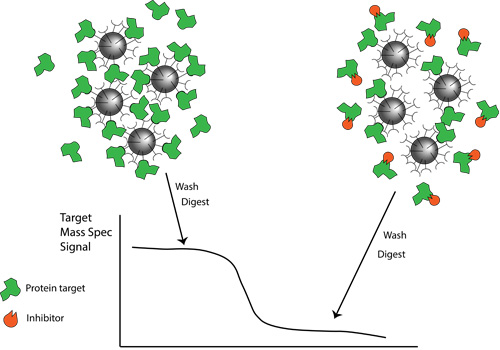
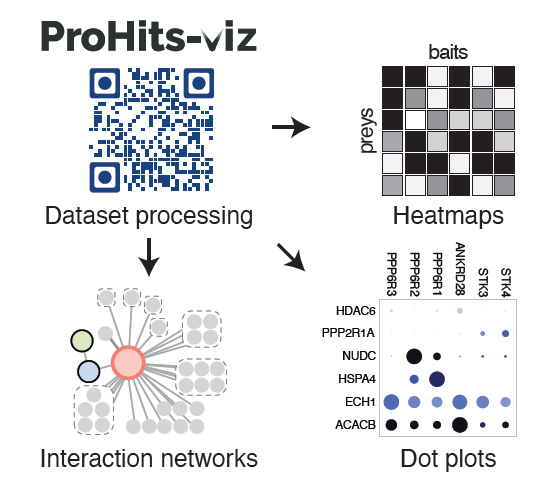
The NBCC provides full support for your proteomics needs from initial study concept to manuscript preparation and data submission to public repositories. The facility uses bottom-up proteomics (shotgun sequencing) to identify peptides using mass spectrometry and then matches them back to known proteins. The facility has developed and validated a first-in-class pipeline for protein-protein interaction studies that culminates in publication-ready visualizations. In addition, we provide advanced instrumentation and in-depth expertise for differential proteome abundance and chemical proteomics studies.